I don’t know if you have ever heard of the lowest common ancestor problem in graph theory and computer science but it’s actually pretty simple. As its name says, it consists of finding the common ancestor for two different nodes which has the lowest level possible in the tree/graph.
Even though it is normally defined for only two nodes given it can easily be extended for a set of nodes with an arbitrary size. This is a quite common scenario that can be found across multiple fields and **taxonomy **is one of them.
The reason I’m talking about all this is because today I ran into the need to make use of such algorithm as part of some improvements in our metagenomics MG7 method. After doing some research looking for existing solutions, I came to the conclusion that I should implement my own, I couldn’t find any applicable implementation that was thought for more than just two nodes.
Ok, but let’s get into detail and see my algorithm:
We start from a set of nodes with an arbitrary length -4 in this sample, which are spread through the taxonomy tree:
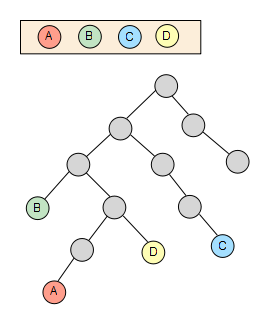
We fetch then the first node from the set and calculate its whole ancestor list to the main root of the taxonomy.
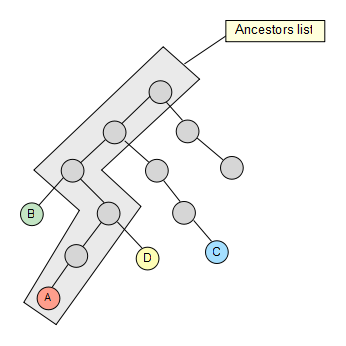
Now that we have the list, we take the second node of the set and check if it’s contained in it, if not, we keep going up through its ancestors until we find a hit. Once the hit has been found, we get rid of the previous elements in the list (if any) so that they are not taken into account for the next iterations in the algorithm.
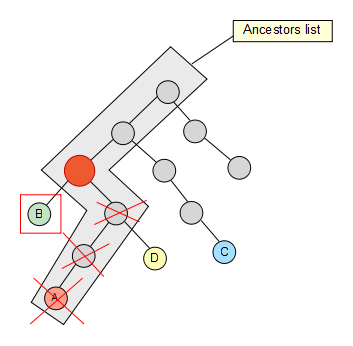
We keep going trough our node set, and C also removes some elements of the list…
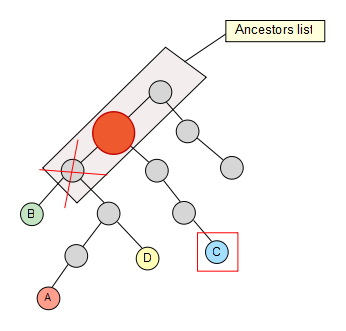
Finally we reach the last node of our set, but no element is removed from our list as a result.
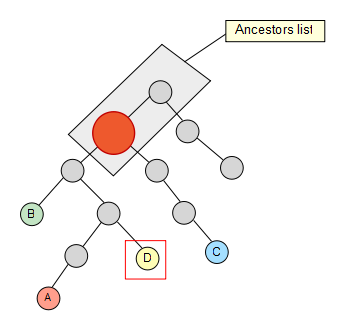
The last thing we have to do is simply get the first element of the resulting list and there we have our lowest common ancestor!
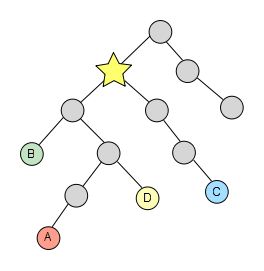
This algorithm is encapsulated in the class TaxonomyAlgo, specifically in the static method lowestCommonAncestor() that expects a list of NCBITaxonNode as parameter and returns their LCA node.
You can also check the class LowestCommonAncestorTest where a simple test program that makes use of this method is implemented.
This program expects as parameters:
- Bio4j DB folder
- An arbitrary number of NCBI taxonomy IDs representing the node set
The Scientific name and the NCBI tax ID of the LCA are printed in the console as result.
Enjoy!
comments
-
Paul Agapow Oddly enough, I had to solve this exact problem a few years ago (to see how much of a tree is left after an extinction, for calculating the biodiversity impact) and then just a few weeks ago (but for the unrooted case). Both times I was sure this had to be a solved problem, but there were no obvious solution out there.
- Pablo Pareja Hi Paul, I was also quite surprised there wasn’t any ‘official’/obvious solution for this, specially when I’d say it’s quite a common problem. Now that you mention it, I think I’ll extend the implementation for the unrooted case as well. By the way, just out of curiosity, what kind of solution did you come up with in the end?
-
Victor de Jager Hi Pablo, interesting post. I solved a very similar problem a few years ago using an early version of the ETE toolkit. http://ete.cgenomics.org/ It’s a well documented with plenty of examples.
- ppareja Hi Victor, Thanks for the link. Just a quick question, is it open-source?
-
Jaime Hi, You may be interested in this python script based on the ETE library: https://github.com/jhcepas/ncbi_taxonomy BTW, ETE is free software
-
Miguel The LCA problem is closely related to the Range Minimum Query problem in graph theory. Working on metagenomics I had to implement a fast algorithm to search for LCA of an arbitrary number of leafs in a taxonomic tree. Given that the tree is always the same, you can pre-process it for fast searches. I ended up implemented the Sparse table algorithm for RMQ explained here: [](http://community.topcoder.com/tc?module=Static&d1=tutorials&d2=lowestCommonAncestor) You say in your post that you couldn’t find any solution out there for more than 2 nodes. The reason is simple: the LCA of N nodes can be decomposed to N-1 times the LCAs of 2 nodes (for example, the LCA of 3 nodes is the LCA of one of them and the LCA of the other 2).
- ppareja Hi Miguel, Thanks for the link ;) In my case though I didn’t want to do any pre-processing on purpose. Having everything stored as a graph gives you a great advantage both in terms of speed and scalability and I didn’t want to throw that away. On the other hand this sort of algorithm is one that could be applied to other sub-graphs of Bio4j, not only the taxonomy tree, so once you implement it in this way it’d be trivial to adapt it to other such cases. I know that the problem can be decomposed so that you end up with a set of 2-nodes problems, what I meant however was that I expected to find algorithms for this problem with some sort of specific optimizations when dealing with a big set of nodes, not only two. For example somehow not passing again through nodes already visited, which will happen when you do decomposing the problem in “isolated” pairs of nodes.